CKD Management

Thickened glomerular basement membranes of diabetic nephropathy as seen on electron microscopy. (Image courtesy of Arkana Laboratories).
CKD MANAGEMENT
Chronic kidney disease clinic is one of the three main settings during which you will see patients. It’s also a vitally important setting because you can alter the course of renal course of a patient for years. Reduce the rate of renal decline by 1% a year for 10 years, and you have potentially kept them off dialysis for a decade. It’s surprising how many different areas you must address in a a CKD clinic visit — eleven to be exact, but if you structure the visit properly and know your plan of action mmediately, you can address all of them even in a short 15 minute follow up visit.
CONSTRUCT YOUR APPROACH
Below are the components that you should address. Look at this overview briefly and we’ll dive into each item individually. Correctly formatting a clinic note is important for a couple of reasons. Firstly, it provides a good framework for your evaluation and treatment for a patient. Secondly, it’s important for you to correctly document your thought process and management decisions both to you, 6 months from now, and to other providers. Therefore, a well-structured clinic note is the goal with the hope and expectation that quality patient care follows.
GCA classification: (grade, cause, albuminuria). Note the trend of eGFR
Metabolic Acidosis
Hyperkalemia
Mineral and Bone Disease
Hypertension
Anemia
Dyslipidemia
Diet Modification
Vaccinations
Education
Access Planning and Care Coordination for Renal Replacement Therapy
GCA Classification
Knowing the etiology, CKD stage, proteinuria, current eGFR, and rate of decline takes up about half a line in a note, but immediately gives an enormous amount of information and frames the decisions made in the rest of the note. The table below outlines how you classify CKD stage and albuminuria (table 1). Per KDIGO guidelines, the CKD-EPI equation is the estimating equation of choice. Many labs, however, still use the MDRD equation. Creatinine clearance (CrCl), estimated from the Cockroft-Gault equation was the first major estimating equation. It’s a relic of the past for renal function estimation, albeit still used for drug dosing in the same way the imperial system is used rather than the metric in our country.
First, you must diagnose CKD for the patient. CKD is defined as abnormalities of kidney structure or function which are present for >3 months, with implications for health. This is a broad definition which means that anything such as albuminuria, renal tubular acidosis, abnormal kidney biopsy findings, history of kidney transplant, structural abnormalities on imaging, urine sediment abnormalities, or GFR < 60 mL/min/1.73m2
CKD is broken down into stages, which are called G-stages and thus have the nomenclature shown below.
G1: GFR >90 mL/min/1.73m2
G2: GFR 60-89 mL/min/1.73m2
G3: GFR 30-59 mL/min/1.73m2
G4: GFR 15-29 mL/min/1.73m2
G5: GFR <15 mL/min/1.73m2
CKD is also broken down into stages of albuminuria, based on albumin-to-creatinine ratios as follows:
A1: ACR <30 mg/g
A2: ACR 30-299 mg/g
A3: ACR at least 300mg/g
DIAGNOSE THE CAUSE OF CKD
After knowing this information, you move onto the etiology of renal failure. This is one of the trickier parts of a CKD clinic visit, because you have to put together a wide variety of data to arrive at a particular etiology. The reason that knowing an etiology is important is because you need to be able to predict the future and you need to know what treatment is available to help a patient retain kidney function. For instance, you don’t want to miss ANCA vasculitis or miss a diagnosis of polycystic kidney disease in a young patient. In both of these cases, we have drugs that will change outcomes. Don’t be lazy and diagnose everyone with diabetic nephropathy or hypertensive nephrosclerosis. Even worse, avoid providing no etiology at all.
How do you arrive at a diagnosis? We can’t perform kidney biopsies on every patient. That would place too many patients at risk for biopsy complications. In addition, something like cardiorenal syndrome doesn't leave a unique fingerprint on renal biopsy. Finding the etiology of CKD is difficult because are so many different things that can cause the kidney’s to fail. Even more, presentations to something as simple as diabetic nephropathy can vary widely and even convince seasoned nephrologists that membranous nephropathy or focal segmental glomerulosclerosis (FSGS) is present.
What we need is a tool that will help us sift through the possible diagnoses? We need something that provides minimal risk to the patient while helping us arrive at an etiology of renal disease even if the renal disease is not explicitly diagnosed with a renal biopsy. This tool just so happens to be Baysian inference. In a nutshell, Baysian inference describes a type of thinking in which you update the probability for a particular CKD etiology continuously as you gather more data. Imagine this scenario — a human being with decreased kidney function. If that is all of the information you know, the the correct diagnosis could essentially be anything. Once you start gathering information, though, you can narrow down the diagnosis. Perhaps the patient has diabetes with a HbA1c of 11% and has had similar lab values for the past 5 years. Perhaps they also have uncontrolled blood pressure and use ibuprofen every day. The eGFR has dropped steadily over this time period. Suddenly, you have gained useful information that helps narrow down potential causes of CKD for this patient. This type of thinking also allows us to make decisions in the setting of incomplete data — the world in which nephrologists often live. Like it was mentioned above, we don’t have kidney biopsy results on every patient, but with Bayesian inference, we can give the patient a reasonable explanation for reduced renal function.
How to start with Bayesian inference — begin with a list of renal disease etiologies that require a significant exposure in order to have one. For instance, it’s difficult to have lead nephropathy without significant exposure to lead. Likewise, diabetic nephropathy without a diagnosis of diabetes is not possible. Needless to say, it’s time-consuming to simply ask all of these yes or no questions during an office visit. Time is better spent explaining the patient’s renal disease to them and your plan for improving their health. Take the form below and include it to the patient in your pre-visit questionnaire. If the patient answers yes to any of these screening questions, go into more detail on them during your office visit.

Screening questions for new CKD patients. Include these on the packet that you mail to the patient. They can fill it out at home and you can quickly view it and ask more detailed questions about positive histories/exposures.
Does the patient have diabetic nephropathy? Begin with a urine albumin-to-creatinine ratio, UA, and HbA1c. Even though diabetic nephropathy is common, it has proven to be a difficult entity to diagnose correctly. The classic presentation is a diabetic patient who develops proteinuria followed by a reduction in eGFR. This is changing over time, however. The phenotype of diabetic nephropathy is shifting to one with less proteinuria and a more predominant reduction in eGFR (Afkarian 2016). To complicate matters further, patients with diabetic nephropathy can have even have hematuria. A study (Matsuma 1994) highlights this with the finding of hematuria in 26 out of 154 patients. 10 out of the 26 with hematuria had IgA nephropathy. The 16 hematuric patients with only diabetic nephropathy had advanced diffuse diabetic glomerular lesions and higher serum creatinine. Ask about history of diabetic retinopathy. The positive and negative predictive values of diabetic retinopathy to detect diabetic nephropathy is 0.72 and 0.69. Possibly the most helpful piece of information is past HbA1c values. Renal damage rarely occurs when postprandial blood glucose levels are <200mg/dL and HbA1c is <7.5-8% (Nosadini 2004); in patients from the ARIC study, adults with DM have hazard ratios for development of diabetic nephropathy over 6 years of 1.4, 2.5, and 3.7 for HbA1c of 6-7%, 7-8%, and >8%, respectively when compared to those with HbA1c of <6% (Bash 2008).
Does a patient have hypertensive nephrosclerosis. This is a common etiology and it is convenient to peg a patient’s CKD on this. 30% of patients reaching ESRD each year receive this diagnosis and this seems questionable. In the Multiple Risk Factor Intervention Trial (MRFIT), the seven year incidence of a doubling of serum Cr was less than 0.2%. In the Hypertension detection and Follow-Up Program, the five year incidence of more than a 25% rise in plasma Cr to above 2.0 was 1.3-1.4% (Freedman 1995). Renal function is typically rock solid in hypertensive nephrosclerosis. It is possible that patients do only have hypertension as a clinical risk factor, but also have underlying genetic factors which place them at risk. The MYHY risk phenotype is associated with non diabetic kidney disease in African Americans and it places them at a 20x greater risk for ESRD from hypertension as compared to European Americans. This is not to mention APOL1 mutations which cause FSGS and proteinuria. Be cautious about simply labeling a middle-aged patient with an eGFR of 20 with HTN controlled with only lisinopril 20mg QD as only having “hypertensive nephrosclerosis.” Hypertensive nephrosclerosis is a heterogenous disease likely includes both FSGS and non-FSGS glomerular diseases, recognized malignant hypertension, renal artery stenosis, and cholesterol emboli syndrome (Freedman 2008).
What about the black box diagnosis of tubulointerstitial disease? The list of things that can cause this type of renal failure is impossibly long. General characterteristics are helpful though. Proteinuria is typically <1g/day, hematuria in uncommon. Even if you did get a renal biopsy, the findings would be non-specific as to the cause of interstitial fibrosis. One possibly distinguishing factor could be the relatively worse anemia seen in chronic interstitial fibrosis as erythropoietin-producing cells are affected early (CCN). Taking a detailed history, or even better, this useful history sheet, is the best way to find the cause. Get rid of the cause and you can stabilize renal function.

An impoosibly long list of etiologies of renal failure — this time from chronic interstitial nephritis. Fortunately, these etiolgogies are again exposure-based and so it’s not out of the question to find an etiology if you look hard enough. The above table is modified from Clinical Comprehensive Nephrology 5e, p746 (Johnson 2014).
What about polycystic kidney disease? Get a renal ultrasound and look for these findings to diagnose polycystic kidney disease:
⁃ positive family history in adults, but unknown genotype (PKD1 or PKD2):
15-39yo: at least 3 unilateral or bilateral cysts
40-59yo: at least 2 cysts in each kidney
60yo or older: at least 4 cysts in each kidney
⁃ at risk for type 1 ADPKD
15-30yo: at least 2 unilateral or bilateral cysts
30-59yo: 2 cysts in each kidney
60yo or older: at least 4 cysts in each kidney
⁃ at risk for type 2 ADPKD
testing for the known mutation is more definitive and may be more cost effective than ultrasound. if genetic testing is not available or less desirable, criteria for at risk patients with unknown phenotype may be used
⁃ if negative family history
most likely it is that the affected parent has died without a diagnosis or is alive with a mild form of the disease. in this case, there is no definitive number of cysts that provides an unequivocal diagnosis of ADPKD, but the diagnosis could be suspected in the presence of the arbitrary number of 10 or more cysts in each kidney and the absence of findings suggestive of a different renal cystic disease , particularly if liver cysts are also present
At this point, you likely have a reasonable explanation for why the patient has CKD, or at least a brief differential diagnosis. It’s possible that after questioning, an etiology is still unknown. This is where initial testing comes into play. Broadly speaking, there are only 3 ways that the kidney can fail — things happening before the kidney, things happening after the kidney, and things happening within the kidney itself. Doing initial testing for all of these is simple, appropriate, and can quickly determine the subset of advanced tests to order, if needed. These tests are, current BMP and trend of prior BMPs, UA, urine microalbumin/creatinine ratio, and renal ultrasound are all that are needed for initial testing to determine the majority of etiologies. At this point, you likely understand the cause of CKD in 90% of the cases. You understand the pathophysiology of their kidney(s) and you can proceed to manage their kidney disease.
Quantify Albuminuria
Why should we quantify proteinuria? On the end of diagnosis, it’s important to stratify patients into nephrotic range proteinuria, non-nephrotic range proteinuria, and those with normal protein excretion. On the therapeutic side of things, we can preserve renal function by reducing proteinuria in certain situations. It’s a fact that the degree of proteinuria is correlated with renal outcomes. The worse your proteinuria is, the worse the prognosis. What is not a fact, and it actually a somewhat controversial topic, is if proteinuria is an appropriate therapeutic target.
It feels intuitively wrong to think that reducing proteinuria may not preserve renal function. Protein in the renal tubules is taken up by proximal tubule cells. Partial degradation of albumin produces antigenic peptides that are then released to the tubulointerstial space where they upregulate inflammatory and fibrotic pathways. How could reducing this not improve renal outcomes? It is sometimes argued that it is difficult to parse out the individual effects of ACE/ARB renoprotection from proteinuria reduction vs ACE/ARB renoprotection from benefits outside of proteinuria reduction alone. The RENAAL trial showed that ARBs result in renoprotection. Across all participants taking an ARB, albuminuria was reduced by 28%, but this varied among individual participants, with some having a >60% reduction in proteinuria and others actually increasing albuminuria. In addition other trials titrating ACE/ARB doses to target a certain reduction in proteinuria might actually be studying the effects of optimal vs suboptimal doses of ACE or ABS. In this view, our use of ACE/ARBs may need to take a shift more like statin use. Rather than targeting certain LDL levels, it is now recommended to use a certain statin (low, moderate, high) intensity for particular patients. Overall, it seems more appropriate to recommend maximizing ACE/ARB use rather than recommending a certain albuminuria target.
Is is better to quantify proteinuria or albuminuria? 24h urine collections are the most accurate way to quantify urine protein and this should be the test of choice, for example, for initiating immunosuppressive therapy for membranous nephropathy. For our more routine CKD patients though, this is a cumbersome approach and so protein-to-creatinine ratios or albumin-to-creatinine ratios should be used. The 2012 KDIGO guidelines for Evaluation and Management of CKD note that albumin-to-creatinine ratio should be used rather than protein-to-creatinine ratio. I’ve heard the argument that since the protein-to-creatinine ratio is less expensive, it should be initially used. This is true, but it is more important to improve patient outcomes, both from a medical standpoint as well as a cost-effectiveness standpoint. Firstly, notable clinically-relevant changes in albuminuria can occur in the absence of large changes in proteinuria. This point is different than albuminuria as a therapeutic target. The AASK trial did show that changes even in non-nephrotic range proteinuria were associated with clinically-relevant changes. Even if we can’t provide renoprotection by reducing proteinuria, it’s important to know what is happening in our patients. The ratio of albumin to urine total protein is about 0.2 when the total protein is <100mg/g creatinine. It is 0.5 when the urine total protein is 200ng/g, and 0.8 when the urine total protein exceeds 1000mg/g (Shihabi 1991). Because of this, there can be an increase in albuminuria in the absence of a significant increase in urine total protein, especially at lower levels of albuminuria. Remember the AASK trial mentioned above? Two-thirds of the patients had a UPCR of 0.22 or less which probably meant that they had albuminuria of maybe 80mg day max?? If someones UPCR increased from 0.22 to 0.26, few would bat an eye, but this could actually mean that the patient had a doubling of albuminuria which was barely noticeable. In those with increasing albuminuria, it may be useful to push harder for a reduction in sodium intake.
But what about the cost? It is argued that because of the lower testing cost, UPCR rather than ACR should be used. It’s difficult to actually figure out how much these tests cost, but even if the albumin-to-creatinine ratio was $15 more per test, but you pushed the need for dialysis a year out farther for one patient a year, you could perform 4,533 urine albumin-to-creatinine ratios per year and break even from a cost-benefit analysis since a year of in-center dialysis costs $68,000 a year.
Day-to-day variability does occur with proteinuria. A great paper (Naresh 2013) looked at day-to-day variability in albumin-to-creatinne ratios and found the following table of expected variability in day-to-day testing (table below). With each level of albuminuria, if the albuminuria falls outside of the expected variability, then a change in proteinuria has occurred. I think that the take-home point from all of this is that it’s important to push ACE/ARBs to the doses used in clinical trials firstly. Specifically targeting a reduction in sodium intake is difficult to actually address in clinic patients (more difficult than just saying “reduce sodium intake”). In patients with increasing albuminuria, it’s likely useful to specifically address this, even as far as getting 24h urine collections for sodium intake.

Day-to-day variability in albuminura as shown on urine albumin-to-creatinine ratios (ACR). The 24h urine albumin excretion refers to albumin excretion based on this 24h test. Baseline ACR refers to a UCR taken on the day of the 24h urine collection. The single repeat test shows the expected range of ACR if the test was repeated.
Manage Hyperkalemia
Hyperkalemia is a daily struggle. The question is how to best deal with it. Choices include lifestyle modifications, diuretics, potassium binders, sodium bicarbonate, and dialysis. A stepwise approach to this is useful so that that potassium level of 5.2mmol/L doesn’t turn into a K+ of 5.9mmol/L by the next visit. Studies ACE and ARB use obviously must address this common issue and provide a good framework for managing this common issue. In the NEPHRON-D study (Fried 2013) has a nice progressive framework for managing hyperkalemia. For mild hyperkalemia between 5.0-5.5mmol/L, they discontinued medications that could lead to hyperkalema and instituted a low potassium diet. For patients with potassium >5.5mmol/L, they added conservative treatment to enhance potassium excretion or potassium redistribution such as the addition or enhanced dosing of diuretics of volume depletion was not a concern. Alkali therapy, increased sodium diet was also instituted if volume overload or hypertension was not a concern as well as low dose sodium polystyrene (15-30g 3 times weekly). If the potassium level was 6.0-6.5mmol/L, then an ECG was performed. If there were no ECG changes, then 60g sodium polystyrene was given. If there were ECG changes or if the potassium was >6.5mmol/L, then acute treatment for hyperkalemia was performed. Of note, the rates of colonic necrosis from sorbitol are estimated to be 0.27% to 1.8% (Fried 2017; Gerstman 1992).
So what about giving sodium bicarbonate therapy for hyperkalemia? Does it work well? It was used in the NEPHRON-D study. UpToDate notes that there are no strong clinical data regarding its effectiveness. Let’s review the relevant renal physiology of potassium excretion with sodium bicarbonate admiinistration outside of overt metabolic acidosis. In general, alkalemia stimulates distal nephron potassium secretion and urinary potassium excretion. elevated intracellular pH decreased the activity of ENaC, ROMK, and BK channels. ENaC abundance is increased when luminal or basolateral bicarbonate is elevated. Moreover, in the case of acute metabolic alkaosis, there is inhibition of factional sodium bicarbonate and fluid reabsorption in the proximal tubule, leading to increased distal delivery of sodium and bicarbonate and enhanced fluid flow. In addition, increased luminal delivery of bicarbonate as a non chloride anion stimulates a component of distal potassium secretion because of the reduced luminal chloride concentration (Aronson 2011).
All in all, for patients with a serum bicarb level <23mmol/L and hyperkalemia, institution of sodium bicarbonate tabs is reasonable.
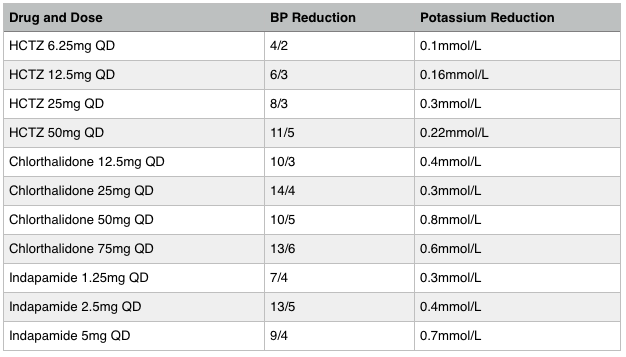
What about diuretics? The level of renal function, blood pressure, volume status will mainly decide if you use thiazide or loop diuretics. I think it is splitting hairs to decide what type of loop diuretic you should use, but keep in mind that thiazide diuretics don’t work well at eGFRs <30. In this case use loop diuretics. If the eGFR is >30 though, there may be some benefit of choosing some thiazide diuretics above others. Indapamide lowers K+ the most, followed by chlorthalidone and lastly, HCTZ. Indapamide 1.25mg lowers K+ more than 1mmol/L. Chlorthalidone lowers potassium by ~0.2mmol/L as shown in the ALLHAT Trial. In the event of hyperklaemia, such as possibly diabetics with hyporenemic hypoaldosteronism, use indapamide rather than HCTZ.
In healthy adults, potassium intake is around 4700mg per day. In patients with CKD, reduction of potassium to 3000mg per day is recommended for those with eGFR <30. Unfortunately, most healthy foods are high in potassium, including those high in fiber. The amount of potassium in certain foods can drastically reduce the amount of potassium contained. For instance, boiling white potatoes can reduce the amount of potassium by 50% (Cupisti 2018). More on potassium management and recipes for CKD will come in the future.
If all of the above therapies fail, cation exchangers such as patiromer or zirconium cyclosilicate can be useful.
Treat Metabolic Acidosis
Serum bicarb levels need to stay in the 23-29mmol/L range. Patients with CKD4 with bicarb levels 16-20mmol/L who were treated had rate of decline of renal function 1.9mg/dL/year vs 5.9mg/dL/year and, over a 2 year period Brito-Ashurst 2009), had a low risk of ESRD (6.5% vs 33%). the data for bone health improvement are not as impressive (21 HD patients) (Lefebvre 1989). it improves muscle strength (Abramowitz 2013). and increases lean muscle mass (de Bristo-Ashurst 2009). Start sodium bicarbonate at dose of 0.5-1.0 mEq/kg/day. Sodium citrate causes less boating, but it should not be used in patents taking aluminum-containing antacids.
Manage Mineral and Bone Disease
KDIGO published an update for CKD-MBD in 2017 and this is, in addition to UpToDate is where the following information comes from. Hypercalcemia should be avoided, but mild asymptomatic hypocalcemia in the setting of calcimemetic treatment can be tolerated in an attempt to avoid inappropriate calcium loading. Remember to correct calcium for albumin (add 0.8mg/dL to total calcium for every g/dL that serum albumin is below 4.0g/dL).
If a patient with CKD 3a-5 develops overt hyperphosphatemia, dietary changes should be undertaken to reduce phosphorous intake. Restriction of daily phosphate intake to 900mg a day should be performed as long as this does not compromise nutritional status. . Remember that around 40-60% of animal-based phosphate is absorbed through the GI tract, but only around 20-50% of plant-based phosphate is absorbed. Phosphorous-containing preservatives in food are very highly absorbed and avoidance of these should be recommended. If the serum phosphorous is still elevated after dietary modification is properly undertaken, then phosphate binders can be prescribed. Different binders have different strengths in regards to phosphorous binding (see the table below). This is a point of contention among experts, though. Since there have been no randomized trials have shown a clinically relevant benefit with phosphate binders in non-dialysis CKD patients, some experts have not recommended them for this population. If they are prescribed, though, non-calcium containing binders should be used as calcium-containing binders lead to vascular calcification. Aluminum-hydroxide should not be used long-term.
Treat vitamin D deficiency. Aim to keep the vitD level between 20-40 ng/mL. If the level is <12ng/mL, give ergocalciferol 50,000 IU weekly for 6-8 weeks and then follow with 800 IU cholecalciferol daily afterwards. If the vitD level is 12-20 ng/mL, the give 800-1000 IU daily. If the level is 20-30 ng/mL, then give 600-800 IU cholecalciferol daily. Per KDIGO guidelines, vitD levels chould be checked and monitored with a frequency appropriate for the degree of deficiency and repletion regimen.
The optimal PTH level in G3a-G5 patients not on dialysis, the optimal PTH level is unknown. If a CKD patient has either vitD deficiency or hyperphosphatemia, the treatments above should be undertaken and they can lower PTH. The real question for management of high PTH levels are if calcitriol should be given. In the setting of high PTH levels with normal phosphorous and repleted vitD levels, the UpToDate authors suggest giving calcitriol 0.25mcg three times weekly if the PTH level is 2.3-3.0 times the upper limit of normal (150-200 pg/mL for an assay with an upper limit of normal of 65 pg/mL) with a final dose titrated to achieve a PTH level of <150 pg/mL. The KDIGO guidelines say that calcitriol should not be routinely used and that it should be used in the situation described above for CKD 4-5 patients.
Dual-energy x-ray absorptiometry (DXA) scans have been an evolving point in CKD over the past decade. The 2017 KDIGO update stated that DXA scans can be performed in CKD 3a-5D if the results will impact treatment decisions.
Bone biopsy is infrequently performed for managment of CKD-MBD, but may be useful in a few situations. There is a nice UpToDate article on this.

Phosphate binder use. In general, reduce the use of calcium-containing binders. Aluminum hydroxide should only be used for very short durations due to risk of aluminum accumulation.
Treat Hypertension
In general, all CKD patients should be treated to a goal BP of <130/80mmHg. The AASK trial, MDRD study, and the SPRINT Trial address this question in CKD patients. There are two outcomes that should be considered for CKD patients -- does a blood pressure goal of <130/80 protect renal function over time and does it reduce mortality. At the end of the original AASK trial (published 2002), intensive blood pressure reduction did not result in reduction in eGFR decline. After the original AASK trial, a 5 year (2002-2007) cohort study of AASK participants took place. Again, there was no reduction in eGFR decline from intensive BP control. However, when participants with UPCR > 0.22 (median proteinuria in these patients was 1000mg/day) were examined, they actually did have a reduction in eGFR decline (HR for progression of CKD was 0.73 for intensive BP control. The MDRD study was published in 1993. After this point, patients were passively followed for 7 years. Those with intensive BP control (goal equivalent to 125/75) were less likely to experience renal failure or death. Subgroup analysis revealed, though, that the benefit of intensive BP control extended only to those with at least 1g/day proteinuria. The SPRINT trial showed that intensive blood pressure reduction did not result in reduction in eGFR decline (intensive BP reduction alternatively did have increased risk of 30% or greater decline in eGFR), but it did show us that intensive BP reduction did significantly lower all-cause mortality. Because of these studies, a BP goal of 130/80 is reasonable for all patients with CKD. This also the recommendation by the American Heart Association. This would provide renoprotection for those with proteinuria >1g/day and reduction in all-cause mortality for everyone with CKD. In patients who are 75 years or older with more of an isolated systolic hypertension picture, or a low diastolic blood pressure, it’s best to individualize blood pressure targets and loosen SBP targets to 135-140. In patients who are very frail or very elderly, individualize blood pressure targets more and firstly, do no harm when instituting blood pressure medications. In these individuals, a broken hip due to a fall from orthostatic hypotension would likely be a worse outcome than a systolic blood pressure that is slightly on the higher side.
ACE inhibitors — which are the best? Lisinopril, benazepril, and ramipril all have once-daily dosing and are $2-6 per month. Lisinopril is the cheapest and so I think it’s the best one. These are not medications worth spending lots of money on. If someone is on benazepril or ramipril, I’ll leave them on it. Enalapril and quinapril don’t last 24h which means they will either have to take them twice daily or have suboptimal blood pressure control if someone prescribes them once daily. I’ll switch these medications to lisinopril. There are no mortality or renoprotection differences between ACE inhibitors.
ARBs — losartan is the only one that is less than $10 per month. If you’re using an ARB, prescribe this one and don’t look back. One last note, a recent study showed that ACE inhibitors and ARBs have equivalent outcomes. Because of this, the general recommendation for ACE or ARB use stands.
Dyhydropyridine CCBs — Amlodipine is better than nifedipine. Fewer patients have high nighttime BP (39%) with amlodipine as compared with BID nifedipine SR (71%). Perfect compliance is 98% with amlodpine and 87% with BID nifedipine. This is an easy choice. Amlodpine 6.7mg/day lowers BP on average 21/15mmHg (Cross 1993 A multicentre study of the safety and efficacy of amlodipine in mild to moderate hypertension).
Duretics — use thiazide diuretics for those without heart failure, decompensated cirrhosis, or eGFR <30. As for the best diuretic, take a look at the table and pick the blood pressure and potassium reduction that suits your patient the best. Do note that chlorthalidone and indapamide have longer duration than HCTZ and are more preferable agents. If the eGFR is <30, use loop diuretics, and pick lasix or bumetanide. Be sure to dose twice daily since diuresis does not last all day.
A 4th agent — what do you use for blood pressure control when you are maximized on ACE (or ARB), CCB, and a diuretic? Spironolactone is the best 4th agent. Patients in the ASCOT Trial who received spironolactone (mean 50mg) had a subsequent drop in blood pressure from 156/85 to a fall of 22/10mmHg. Hyperkalemia to >5.5 occurred in 4%. In a randomized, double-blind crossover trial (PATHWAY-2 Trial), spironolactone 25-50mg reduced SBP by 10mmHg whereas doxazosin and bisoprolol reduced it by 5mmHg and 6mmHg, respectively. Absolute contraindications for spironolactone include eGFR <30, K+ >5.5 and diabetics with microalbuminuria (Jain 2009). the microalbuminuria part is there because these patients will be more likely to have hyperkalemia. Is it bad to use spironolactone with ACE inhibitor (dual RAAS blockade)…no (Jain 2009). After you can’t control blood pressure with 4 agents, individualize blood pressure treatment based on side effect profiles of each agent.
Beta blockers — What role do beta blockers have? Aside from obvious uses such as in patients with coronary artery disease or AFib, they mainly play a role as a 5th or 6th agent, along with doxazosin, clonidine, hydralazine, ect. I believe I would personally start them as a 5th agent and then use doxazosin, clonidine, or hydralazine as a 6th agent. Beta-blockers probably make little or no difference in the number of deaths among people on treatment for high blood pressure. This effect appears to be similar to that of diuretics and renin-angiotensin system inhibitors, but beta-blockers are probably not as good at preventing deaths from high blood pressure as calcium-channel blockers.(Wiysonage 2017). People given beta-blockers are more likely to have side effects and stop treatment than people taking renin-angiotensin system inhibitors, but there may be little or no difference in side effects between beta-blockers and diuretics or calcium-channel blockers (Wiysonage 2017). beta-1 selective blockers lowered BP by an average of -10/-8 mmHg and reduced heart rate by 11 beats per minute as compared to placebo. The effect of beta-1 blockers at peak hours, -12/-9 mmHg, was greater than the reduction at trough hours, -8/-7 mmHg. Beta-1 selective blockers lowered BP by a greater magnitude than dual receptor beta-blockers and partial agonist beta-blockers, lowered BP similarly to nonselective beta-blockers. Beta-1 selective blockers lowered SBP by a similar degree and lowered DBP by a greater degree than diuretics, angiotensin converting enzyme inhibitors and angiotensin receptor blockers (Wong 2016). atenolol 100mg QD reduces BP 12/10. metoprolol in total daily dosage ranges of 100-400mg daily reduce BP 9/8 without a dose response curve in this range. lower doses of 25-50mg a day lowered BP to a smaller degree than the does ranges mentioned above. bisoprolol at the starting dose of 5mg once daily reduces BP to the same degree (11/8) as 10mg and 20mg a day (Wong 2016). BB use after a myocardial infarction can use any type of BB unless it has intrinsic sympathomimetic activity such as labetalol. on the other hand, labetalol may have less bradycardia and could be useful in patients who have excessive bradycardia with other agents. dual alpha and beta blockers (labetalol and carvedilol) reduce BP by 4/3. based on indirect comparison with other BB, the BP lowering effect of these types of BB is less than that of beta-1 selective and non-selective BB classes. dual alpha and beta blockers have less BP reduction than thiazides and drugs inhibiting RAS (Wong 2015).
Treat Anemia
Anemia is present if Hb <13 g/dL in men and <12 g/dL in women. The prevalence of anemia (hemoglobin [Hb] <12 g/dL in men and <11 g/dL in women) increased from 1 percent among patients with an estimated GFR (eGFR) of 60 mL/min/1.73 m2 to 9 percent at an eGFR of 30 mL/min/1.73 m2 and to 33 to 67 percent at an eGFR of 15 mL/min/1.73 m2. Monitor annually if CKD3a and twice annually if CKD3b for anemia. If anemia is present, monitor Hb twice annually is CKD3a and every 3 months if CKD3b. If iron TSAT is <20% and ferritin is <100, give PO iron. If the patient has anemia and TSAT is <30 and ferritin is <500, give PO iron. In general, give ferrous sulfate 325mg TID. The number needed to harm is 11 in regards to GI side effects; slow release iron has lower (3.3% vs 6.4%) rates of nausea and epigastric pain than elemental iron (McDiarmid 2002). Some new research has come out which showes that showed that amount of absorbed iron in 120mg QAM and 60mg QPM was no different then 120mg QAM only; iron iron doses >60mg result in higher fractional absorption when dosages are spaced by 48h (Moretti 2015). alternate day dosing is also recommended by another group (stifle 2017). Every other day dosing could be reasonable in mild iron deficiency. Otherwise, use ferrous sulfate 325mg TID. Use IV iron when PO no longer works. What dose of IV iron is best? low dose (50mg iron per week) vs high dose (>400mg per month) has been a debate. a meta-analysis of these two dosing strategies demonstrated no safety difference between the two (Hougan 2018). these studies, however, took place over a long time period and there is significant heterogenity in the studies (Li 2018). in summary, either dosing strategy is ok. ESAs are used for patients with TSAT >25% and ferritin >200. SubQ dosing uses 30% less than IV dosing and so use that. Maintain Hb in the 10.0-11.5 range. It is not definitely clear that ESAs increase the risk of cancer, but the field is still developing and so it’s useful to take e a cautious approach. Use normal protocols if there is no history of cance.r i fhtere is no cancer, but there are major risk factors, consider a conservative Hb target of 10.0. Active cancer: treat corrective causes; upper limit of Hb 10.0; treat under the APPRISE REMS program; monitor and prevent thromboembolic events. past history of cancer: talk to the patient’s oncologist; until cancer is <5 years past cure date, then treat as if active cancer is present; after that point, then consider continuing conservative treatment Hb target.
At what eGFR does anemia typically start?
at an eGFR of 60, 8% of women and 7% of men will have anemia
at an eGFR of 45, 15% of women and 12% of men will have anemia
at an eGFR of 30, 38% of women and 29% of men will have anemia
at an eGFR of 15, 82% of women and 69% of men will have anemia
all of the above data is from Astor 2002 which is from the NHANES study
Treat Hyperlipidemia
The primary finding is hypertriclyceridemia with total cholesterol being normal. Screen new patients with a lipid panel. Give statin or statin/ezetemibe combination for patients >50yo with eGFR <60in patients 18-49yrs, give statin if a patient has known CAD, had DM, prior ischemic stroke, or a 10-year estimated incidence of coronary death or non-fatal MI that is >10%. Give statin for patients with kidney transplants. The SHARP trial (Baigent 2011) showed that statin/ezetemibe combination reduced major atherosclerotic events by 17%. Statins are important to keeping our patients healthy. Fluvastatin 80mg daily, atorvastatin 20mg daily, rosuvastatin 10mg daily, pravastatin 40mg daily, and simvastatin 40mg daily have all ben shown to be beneficial in CKD populations (KDIGO guidelines for lipids) and so give one of these. Do note that simvastatin doses of 20mg/day or greater, when combined with amlodipine, increase the risk for myopathy. As most of my patients are on amlodipine, I shy away from this one and typically prefer atorvastatin.
GIVE APPROPRIATE Vaccinations
Give annual influenza. Patients with stage 4-5 CKD should et polyvalent pneumococcal vaccine unless contraindicated. patients who get pneumococcal vaccination should be offered revaccination within 5 years. If they are naive to this vaccine, give PCV13; after 8 weeks, then give PPSV23; after 5 years give PPSV23; give PPSV23 at the age of 65 and then again every 5 years. If they have already had PPSV23, but not PCV13, then give PCV13 at least 1 year after PPSV23, then give second dose of PPSV23 5 years after first dose of PPSV23 and >8 weeks after PCV13; give another dose of PPSV23 at age 65 and then every 10 years.
References
Abramowitz, M. K. (2017). Bicarbonate balance and prescription in ESRD. Journal of the American Society of Nephrology, 28(3), 726-734.
Afkarian, M., Zelnick, L. R., Hall, Y. N., Heagerty, P. J., Tuttle, K., Weiss, N. S., & De Boer, I. H. (2016). Clinical manifestations of kidney disease among US adults with diabetes, 1988-2014. Jama, 316(6), 602-610.
Appel, L. J., Wright, J. T., Greene, T., Kusek, J. W., Lewis, J. B., Wang, X., ... & Contreras, G. (2008). Long-term effects of renin-angiotensin system–blocking therapy and a low blood pressure goal on progression of hypertensive chronic kidney disease in african americans. Archives of Internal Medicine, 168(8), 832-839.
Aronson, P. S., & Giebisch, G. (2011). Effects of pH on potassium: new explanations for old observations. Journal of the American Society of Nephrology, 22(11), 1981-1989.
Baigent, C., Landray, M. J., Reith, C., Emberson, J., Wheeler, D. C., Tomson, C., ... & Neal, B. (2011). The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. The Lancet, 377(9784), 2181-2192.
Bash, L. D., Selvin, E., Steffes, M., Coresh, J., & Astor, B. C. (2008). Poor glycemic control in diabetes and the risk of incident chronic kidney disease even in the absence of albuminuria and retinopathy: Atherosclerosis Risk in Communities (ARIC) Study. Archives of internal medicine, 168(22), 2440-2447.
Chen, W., & Abramowitz, M. K. (2014). Treatment of metabolic acidosis in patients with CKD. American journal of kidney diseases, 63(2), 311-317.
Dahlöf, B., Sever, P. S., Poulter, N. R., Wedel, H., Beevers, D. G., Caulfield, M., ... & Mehlsen, J. (2005). Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. The Lancet, 366(9489), 895-906.
de Brito-Ashurst, I., Varagunam, M., Raftery, M. J., & Yaqoob, M. M. (2009). Bicarbonate supplementation slows progression of CKD and improves nutritional status. Journal of the American Society of Nephrology, 20(9), 2075-2084.
Freedman, B. I., Iskandar, S. S., & Appel, R. G. (1995). The link between hypertension and nephrosclerosis. American journal of kidney diseases, 25(2), 207-221.
Fried, L. F., Emanuele, N., Zhang, J. H., Brophy, M., Conner, T. A., Duckworth, W., ... & Reilly, R. F. (2013). Combined angiotensin inhibition for the treatment of diabetic nephropathy. New England Journal of Medicine, 369(20), 1892-1903.
Fried, L., Kovesdy, C. P., & Palmer, B. F. (2017). New options for the management of chronic hyperkalemia. Kidney international supplements, 7(3), 164-170.
Gerstman, B. B., Kirkman, R., & Platt, R. (1992). Intestinal necrosis associated with postoperative orally administered sodium polystyrene sulfonate in sorbitol. American journal of kidney diseases, 20(2), 159-161.
Jain, G., Campbell, R. C., & Warnock, D. G. (2009). Mineralocorticoid receptor blockers and chronic kidney disease. Clinical journal of the American Society of Nephrology, 4(10), 1685-1691.
Klahr, S., Levey, A. S., Beck, G. J., Caggiula, A. W., Hunsicker, L., Kusek, J. W., & Striker, G. (1994). The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. New England Journal of Medicine, 330(13), 877-884.
Johnson, R. J., Feehally, J., & Floege, J. (2014). Comprehensive Clinical Nephrology E-Book. Elsevier Health Sciences.
Lefebvre, A., De Vernejoul, M. C., Gueris, J., Goldfarb, B., Graulet, A. M., & Morieux, C. (1989). Optimal correction of acidosis changes progression of dialysis osteodystrophy. Kidney international, 36(6), 1112-1118.
Li, X., & Kshirsagar, A. V. (2018). Rest easy with intravenous iron for dialysis patients?: high dose IV iron safety.
Matsumura, N., Hanatani, M., Nishino, T., Ishihara, K., Kishimoto, T., Tonomura, Y., ... & Dohi, K. (1994). The clinico-pathological significance of hematuria in diabetics. Nihon Jinzo Gakkai Shi, 36(9), 1036-1045.
McDiarmid, T., & Johnson, E. D. (2002). Are any oral iron formulations better tolerated than ferrous sulfate?. Clinical Inquiries, 2002 (MU).
Moretti, D., Goede, J. S., Zeder, C., Jiskra, M., Chatzinakou, V., Tjalsma, H., ... & Zimmermann, M. B. (2015). Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood, The Journal of the American Society of Hematology, 126(17), 1981-1989.
Multiple Risk Factor Intervention Trial Research Group. (1982). Multiple Risk Factor Intervention Trial: risk factor changes and mortality results. Jama, 248, 1465-1477.
Nosadini, R., & Tonolo, G. (2004). Relationship between blood glucose control, pathogenesis and progression of diabetic nephropathy. Journal of the American Society of Nephrology, 15(1 suppl), S1-S5.
Naresh, C. N., Hayen, A., Weening, A., Craig, J. C., & Chadban, S. J. (2013). Day-to-day variability in spot urine albumin-creatinine ratio. American journal of kidney diseases, 62(6), 1095-1101.
Shihabi, Z. K., Konen, J. C., & O'connor, M. L. (1991). Albuminuria vs urinary total protein for detecting chronic renal disorders. Clinical chemistry, 37(5), 621-624.
SPRINT Research Group. (2015). A randomized trial of intensive versus standard blood-pressure control. New England Journal of Medicine, 373(22), 2103-2116.
Stoffel, N. U., Cercamondi, C. I., Brittenham, G., Zeder, C., Geurts-Moespot, A. J., Swinkels, D. W., ... & Zimmermann, M. B. (2017). Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. The Lancet Haematology, 4(11), e524-e533.
Williams, B., MacDonald, T. M., Morant, S., Webb, D. J., Sever, P., McInnes, G., ... & Mackenzie, I. (2015). Spironolactone versus placebo, bisoprolol, and doxazosin to determine the optimal treatment for drug-resistant hypertension (PATHWAY-2): a randomised, double-blind, crossover trial. The Lancet, 386(10008), 2059-2068.
Wiysonge, C. S., Bradley, H. A., Volmink, J., Mayosi, B. M., & Opie, L. H. (2017). Beta‐blockers for hypertension. Cochrane database of systematic reviews, (1).
Wong, G. W., Boyda, H. N., & Wright, J. M. (2016). Blood pressure lowering efficacy of beta‐1 selective beta blockers for primary hypertension. Cochrane Database of Systematic Reviews, (3).
Wong, G. W., Laugerotte, A., & Wright, J. M. (2015). Blood pressure lowering efficacy of dual alpha and beta blockers for primary hypertension. Cochrane Database of Systematic Reviews, (8).
Wright Jr, J. T., Bakris, G., Greene, T., Agodoa, L. Y., Appel, L. J., Charleston, J., ... & Hebert, L. (2002). Effect of blood pressure lowering and antihypertensive drug class on progression of hypertensive kidney disease: results from the AASK trial. Jama, 288(19), 2421-2431.